L’ivermectine (Stromectol) est un antiparasitaire dont l’action repose sur la liaison sélective aux canaux chlore activés par le glutamate présents dans les cellules nerveuses et musculaires des parasites. Cette fixation entraîne une augmentation du flux de chlore, provoquant une hyperpolarisation et une paralysie irréversible. L’ivermectine est active contre la gale, l’onchocercose et certaines strongyloïdoses. Sa biodisponibilité orale est variable, augmentée par la prise alimentaire, et son élimination est principalement fécale via un métabolisme hépatique. Elle ne traverse pas la barrière hémato-encéphalique, ce qui limite les effets neurologiques chez l’homme. Les précautions concernent l’interaction avec les inhibiteurs du CYP3A4, ainsi que les réactions inflammatoires dues à la destruction massive des parasites. Dans les documents de référence, stromectol prix est associé à des protocoles précis adaptés aux différentes infestations, avec une attention particulière sur la sécurité d’emploi en cas d’immunodépression.
Microsoft word - prii-v05
PRILIGY (dapoxetine hydrochloride)
SYNCOPE: Patients on PRILIGY need to be made aware that they could experience syncope at any time with or without prodromal symptoms during their treatment with PRILIGY. Syncope characterized as loss of consciousness has been reported in clinical trials and is considered medicinal product-related. A dose-response relationship is suggested. Prodromal symptoms (such as nausea, dizziness or light-headedness) often preceded the syncope. Patients should be counselled about the importance of maintaining adequate hydration, and about how to recognise and act upon prodromal signs and symptoms (see PRECAUTIONS) to decrease the likelihood of serious injury associated with falls due to loss of consciousness. Patients must not take more than one tablet once every 24 hours due to increased risk. NAME OF THE MEDICINE
Dapoxetine hydrochloride has the following chemical structure:
DESCRIPTION
Dapoxetine hydrochloride belongs to the pharmacotherapeutic group of selective serotonin reuptake inhibitors (SSRIs).
PRILIGY is available as 30 mg tablets. Each PRILIGY 30 mg film-coated tablet contains 30 mg of dapoxetine base (as hydrochloride). Inactive ingredients: lactose, cellulose-microcrystalline, croscarmellose sodium, silica-colloidal anhydrous, magnesium stearate, hypromellose, glycerol triacetate, titanium dioxide, iron oxide black, iron oxide yellow.
Dapoxetine hydrochloride is a white to slightly yellow powder. It is freely soluble in methanol, propylene glycol, some organic solvents (e.g. N, N-dimethylformamide) and slightly soluble in ethanol. The solubility of dapoxetine hydrochloride in aqueous media is a function of the pH (soluble at pH 3.9, sparingly soluble at pH 2.1 and insoluble at pH >7.0).
The chemical name is (+)-(S)-N, N-dimethyl-()-[2-(1-naphthalenyloxy) ethyl]- benzenemethanamine hydrochloride.
PHARMACOLOGY Pharmacodynamics
Dapoxetine inhibits the serotonin transporter. The mechanism of action of dapoxetine in premature ejaculation is presumed to be linked to the inhibition of neuronal reuptake of serotonin and the subsequent potentiation of the neurotransmitter's action at pre- and post-synaptic receptors.
Human ejaculation is primarily mediated by the sympathetic nervous system. The ejaculatory pathway originates from a spinal reflex centre, mediated by the brain stem, which is influenced initially by a number of nuclei in the brain (medial preoptic and paraventricular nuclei). In the rat, dapoxetine inhibits the ejaculatory expulsion reflex by acting at a supraspinal level with the lateral paragigantocellular nucleus (LPGi) as a necessary brain structure for the effect. Post ganglionic sympathetic fibres that innervate the seminal vesicles, vas deferens, prostate, bulbourethral muscles, and bladder neck cause them to contract in a coordinated fashion to achieve ejaculation. Dapoxetine modulates this ejaculatory reflex in rats, causing an increase in pudendal motoneuron reflex discharge (PMRD) latency.
Pharmacokinetics
Dapoxetine is rapidly absorbed with maximum plasma concentrations (Cmax) occurring
approximately 1-2 hours after tablet intake. The absolute oral bioavailability of dapoxetine is approximately 40% (range 15-76%). Following single oral doses of 30 mg and 60 mg in the fasted state, peak plasma concentration of dapoxetine were 297 ng/ml after 1.01 hours, and 498 ng/ml after 1.27 hours, respectively.
Ingestion of a high fat meal modestly reduced the Cmax (by 10%) and modestly increased the
AUC (by 12%) of dapoxetine and slightly delayed the time for dapoxetine to reach peak concentrations; however, the extent of absorption was not affected by consumption of a high fat meal. These changes are not clinically significant. PRILIGY can be taken with or without food.
Greater than 99% of dapoxetine is bound in vitro to human plasma proteins. The active metabolite desmethyl dapoxetine is 98.5% protein bound. Dapoxetine appears to have a rapid distribution with a mean steady state volume of distribution of 162 L. Following intravenous administration in humans, mean estimated initial, intermediate, and terminal half-life values for dapoxetine were 0.10, 2.19, and 19.3 hours respectively.
In vitro studies suggest that dapoxetine is cleared by multiple enzyme systems in the liver and kidneys, primarily CYP2D6, CYP3A4, and flavin monooxygenase 1 (FMO1). Following oral dosing in a clinical study designed to explore the metabolism of 14C-dapoxetine, dapoxetine was extensively metabolised to multiple metabolites primarily through the following biotransformational pathways: N-oxidation, N-demethylation, naphthyl hydroxylation, glucuronidation and sulfation. There was evidence of presystemic first-pass metabolism after oral administration.
Intact dapoxetine and dapoxetine-N-oxide were the major circulating species in the plasma. In vitro functional and binding studies showed that dapoxetine-N-oxide was only weakly active at the serotonin transporter. Additional metabolites include desmethyldapoxetine and didesmethyldapoxetine, which account for less than 3% of the circulating medicinal product-related material. In vitro functional studies indicate that desmethyldapoxetine is equipotent to dapoxetine and didesmethyldapoxetine has approximately 50% of the potency of dapoxetine. The unbound AUC of desmethyldapoxetine is ~40-50% of the free exposure of dapoxetine. The unbound (Cmax) of desmethyldapoxetine is estimated to be 15-20% of dapoxetine Cmax in the
absence of intrinsic or extrinsic factors that may change exposure levels.
The metabolites of dapoxetine were primarily eliminated in the urine as conjugates. Unchanged active substance was not detected in the urine. Dapoxetine has a rapid elimination, as evidenced by a low concentration (less than 5% of peak) 24 hours after dosing. There was minimal accumulation of dapoxetine following daily dosing. The terminal half-life is approximately 19 hours following oral administration. The half-life of desmethyldapoxetine is similar to that of dapoxetine.
Analyses of single dose clinical pharmacology studies using 60 mg dapoxetine indicated no statistically significant differences between Caucasians, Blacks, Hispanics and Asians. A clinical study conducted to compare the pharmacokinetics of dapoxetine in Japanese and Caucasian subjects showed 10% to 20% higher plasma levels (AUC and peak concentration) of dapoxetine in Japanese subjects due to lower body weight. The slightly higher exposure is not expected to have a meaningful clinical effect.
Analyses of a single dose clinical pharmacology study using 60 mg dapoxetine showed that healthy elderly subjects had a slight increase in AUCinf, by approximately 12%, and a mean
terminal half-life of approximately 26 hours. This slightly higher exposure and longer half-life is not expected to have a meaningful clinical effect.
In a single dose clinical pharmacology study using 60 mg dapoxetine, no correlation was noted between creatinine clearance and dapoxetine Cmax or AUCinf in subjects with mild (creatinine
clearance 50 to 80 mL/min), moderate (creatinine clearance 30 to <50 mL/min), and severe (creatinine clearance <30 mL/min) renal impairment. Dapoxetine pharmacokinetics have not been evaluated in patients requiring renal dialysis. Limited data (n=4) in subjects with severe renal impairment showed an approximate 100% increase in AUCinf when compared to those
healthy subjects with no renal impairment (see PRECAUTIONS and DOSAGE AND ADMINSTRATION).
In patients with mild hepatic impairment, unbound Cmax of dapoxetine is decreased by 28% and
unbound AUC is unchanged. The unbound Cmax and AUC of the active fraction (the sum of the
unbound exposure of dapoxetine and desmethyldapoxetine) were decreased by 30% and 5%, respectively. In patients with moderate hepatic impairment, unbound Cmax of dapoxetine is
essentially unchanged (decrease of 3%) and unbound AUC is increased by 66%. The unbound Cmax and AUC of the active fraction were essentially unchanged and doubled, respectively.
In patients with severe hepatic impairment, the unbound Cmax of dapoxetine was decreased by
42% but the unbound AUC was increased by approximately 223%. The Cmax and AUC of the
active fraction had similar changes (decrease of 41% and increase of 241% respectively) (see CONTRAINDICATIONS and DOSAGE AND ADMINISTRATION).
In a single dose clinical pharmacology study using 60 mg dapoxetine, plasma concentrations in poor metabolisers of CYP2D6 were higher than in extensive metabolisers (approximately 31% higher for Cmax and 36% higher for AUCinf) of dapoxetine and 98% higher for Cmax and 161%
higher for AUCinf of desmethyldapoxetine). The active fraction of dapoxetine may be increased
by approximately 46% at Cmax and by approximately 90% for AUC. This increase may result in a
higher incidence and severity of dose dependent adverse events (see DOSAGE AND ADMINISTRATION). The mean terminal half-life in poor metabolisers of CYP2D6 was approximately 22 hours, in comparison to a mean terminal half-life of approximately 19.5 hours, observed in extensive metabolisers of CYP2D6. The safety of dapoxetine in poor metabolisers of CYP2D6 is of particular concern with concomitant administration of other medicinal products that may inhibit the metabolism of dapoxetine such as moderate and potent CYP3A4 inhibitors (see PRECAUTIONS and DOSAGE AND ADMINSTRATION).
Plasma concentrations of dapoxetine and desmethyldapoxetine in CYP2D6 ultrarapid metabolisers are expected to be decreased.CLINICAL TRIALS
The effectiveness of PRILIGY in the treatment of premature ejaculation (PE) was established in four double-blind, placebo-controlled clinical trials, in which a total of 1616 subjects were randomised to PRILIGY 30 mg and 1612 to placebo. All subjects were 18 years of age or older; 99% were <64 years of age. All subjects were heterosexual males, in a stable relationship for at least 6 months, had PE defined by the DSM-IV-TR criteria and in two trials had at least moderate distress or interpersonal difficulty and had an intravaginal ejaculatory latency time (IELT; time from vaginal penetration to the moment of intravaginal ejaculation) of ≤ 2 minutes in a minimum of 75% of evaluable sexual intercourse events during the baseline period. Subjects with controlled hypertension (SBP<180 mmHg, DBP<100 mmHg were included in Phase 3 studies. Subjects with other forms of sexual dysfunction, including erectile dysfunction and those using other forms of pharmacotherapy for the treatment of PE or antidepressants were excluded from all studies.
In all studies, subjects were instructed to administer PRILIGY 1-3 hours prior to anticipated sexual activity and not to take more than one dose per 24 hours. Three of the studies were 12 weeks in duration (C-2002-012, C-2002-013, R096769-PRE-3003), and one study was 24 weeks of treatment followed by a one week assessment of possible withdrawal effects (R096769-PRE-3001).
In the analysis of the pooled studies, mean average IELT at Week 12 (LPOCF) was significantly greater (p<0.001) in the PRILIGY 30 mg (3.1 min) than in the placebo group (1.95 min) (Table 1). These results were similar to those seen in each Phase 3 study, where mean average IELT at Week 12 (LPOCF) was statistically greater for PRILIGY 30 mg compared to the placebo group. In the 12 week US studies that investigated PRILIGY 30 mg (C-2002-012 and C-2002-013), 1.1% of subjects withdrew due to lack of efficacy, 4.8% due to personal reasons, 4.0% due to adverse events, 4.6% due to withdrawal of consent, 6.3% due to lost to follow up, 1.3% due to non-compliance or a protocol violation and 0.6% due to other reasons. In the 12 week Pan-Asian study R096769-PRE-3003 that investigated PRILIGY 30 mg, 1.1% of subjects withdrew due to insufficient response, 9.9% due to personal reasons, 1.7% due to AEs, 0.8% due to lost to follow up, and 5.9% due to other reasons. In the 24 week EU study R096769-PRE-3001 that investigated PRILIGY 30 mg, 6.4% of subjects withdrew due to insufficient response, 14.4% due to personal reasons, 3.9% due to AEs, 5.7% due to lost to follow up and 12.4% due to other reasons.
Table 1: Mean Average IELT§ Results in Phase 3 Placebo-Controlled Studies Pooled Studies Mean Average IELT(SD), Minutes C-2002-012 Mean Average IELT (SD), Minutes C-2002-013 Mean Average IELT(SD), Minutes R096769-PRE-3001 Mean Average IELT(SD), Minutes R096769-PRE-3003
+ Endpoint = Last observation carried forward (LPOCF) to week 12 § Calculated as the treatment group mean from individual subjects' average IELT (Studies C-2002-012, C-2002-013, R096769-PRE-3003) and Week 24 (Study R096769-PRE-3001) *Pooled placebo and Priligy 30 mg groups
In a representative study (R096769-PRE-3001) with the longest treatment duration (24 weeks), 1162 subjects were randomized, 385 to placebo, 388 to PRILIGY 30 mg as needed, and 389 to PRILIGY 60 mg as needed. The mean average IELT at baseline and study endpoint for placebo and 30 mg as needed treatment groups is shown in Figure 1. Increases in mean average IELT at the week 24 endpoint (LPOCF) were statistically significant (p<0.001) in the PRILIGY group versus placebo. The magnitude of IELT prolongation was related to baseline IELT and was variable between individual subjects. The clinical relevance of PRILIGY treatment effects are described below in terms of patient reported response rates.
Figure 1: Study R096769-PRE-3001
Treatment Group: PLACEBO PRILIGY 30 MG PRN
End Point (TRT WK12) = LPOCF to Week 12. End Point (TRT WK24) = LPOCF to Week 24. LPOCF is last post-baseline observation carried forward.
In addition to the primary endpoint of average IELT, meaningful treatment benefit to the patient in the above study was demonstrated using a definition of treatment response consisting of a composite of at least a 2-category increase in control over ejaculation (response options: very poor, poor, fair, good, and very good) plus at least a 1-category decrease in ejaculation-related distress (response options: not at all, a little bit, moderately, quite a bit, and extremely). A greater percentage of subjects responded in the PRILIGY group versus placebo beginning at Week 4 and up to and including Week 24 (this was statistically significant; p=0.003 for dapoxetine 30 mg versus placebo at Week 16, all other comparisons p0.001). Significant decrease in subject distress and significant improvement in subject satisfaction with sexual intercourse were also observed (response options: very poor, poor, fair, good, and very good). Improvements at weeks 12 and 24 (LPOCF) for the key secondary endpoints are presented in Table 2.
Table 2: Percentage of Subjects with Improvement in Key Secondary Endpoints (Study R096769-PRE-3001)
(change 2 in control and -1 in distress)
* p-value <0.001 for PRILIGY versus placebo; LPOCF is last post-baseline observation carried forward Control rated as control over ejaculation: very poor, poor, fair, good, and very good Satisfaction rated as satisfaction with sexual intercourse: very poor, poor, fair, good, and very good Distress rated as distress related to timing of ejaculation: not at all, a little bit, moderately, quite a bit, and extremely
Other secondary patient reported outcome (PRO) endpoints were assessed in the clinical trials including clinical global impression of change in condition, CGIC, a commonly used measure in which patients assess the status of their condition. Patients were asked to compare their premature ejaculation from the start of the study, with response options ranging from much better to much worse. CGIC results by treatment group reported at the end of the above study are shown in Table 3.
Table 3: Results of Clinical Global Impression of Change in Condition at Study
Endpoint (LPOCF*); Study R096769-PRE-3001
* LPOCF is last post-baseline observation carried forward **No Change or Worse includes No Change, Slightly Worse, Worse or Much Worse
At least Slightly Better CGIC response rate (includes Slightly Better, Better and Much Better): Placebo (32%) and Priligy 30 mg (57.7%)
Although PRILIGY 60 mg is not approved, this dose was used to test the withdrawal effects of chronic daily and as needed dosing of PRILIGY in the treatment of premature ejaculation in a placebo-controlled, double-blind, parallel-group study in which 1238 subjects were randomized. Subjects diagnosed with PE based on the DSM-IV-TR without an IELT restriction received placebo or 60 mg PRILIGY either once daily or as needed for 62 days followed by a withdrawal assessment phase of 7 days of additional PRILIGY treatment or placebo. Withdrawal effects after abrupt cessation of therapy were measured using the Discontinuation Emergent Signs and Symptoms (DESS), a clinician-rated instrument that queries for symptoms and signs associated with the discontinuation of serotonin reuptake inhibitor treatment. The DESS specifies a check
list of 43 symptoms that are clinician rated as 1 of 4 categories (i.e., new symptom; old symptom, but worse; old symptom, but improved; and old symptom, but unchanged, or symptom not present. Those symptoms rated by the clinician as "new" or "old, "but worse" are counted as points indicative of a possible withdrawal effect. For each subject, discontinuation syndrome was defined as an increase in the weekly DESS score by at least 4 points from Day 63 to Day 70. In this study, there was no clear evidence of discontinuation (withdrawal) syndrome upon abrupt discontinuation of PRILIGY therapy. Consistent with the lack of discontinuation syndrome based on DESS, adverse event data showed little evidence of withdrawal symptoms. Similar results were seen in a second double-blind clinical trial with a 24-week treatment phase of 30 and 60 mg doses as needed followed by a 1-week withdrawal assessment period.
In the two multidose Phase 3 studies (C-2002-012 and C-2002-013) where the CYP2D6 metaboliser status was identified, a total of 120 poor metabolisers and 1598 extensive metabolisers were enrolled and treated with PRILIGY. No overall differences were seen in efficacy or safety between poor and extensive metabolisers.
INDICATIONS
PRILIGY is indicated for the treatment of premature ejaculation (PE) in men 18 to 64 years of age, who have all of the following: an intravaginal ejaculatory latency time (IELT) of less than two minutes; and
persistent or recurrent ejaculation with minimal sexual stimulation before, on, or shortly
after penetration and before the patient wishes; and
marked personal distress or interpersonal difficulty as a consequence of PE; and
CONTRAINDICATIONS
PRILIGY is contraindicated in patients with known hypersensitivity to dapoxetine hydrochloride or to any of the excipients.
PRILIGY is contraindicated in patients with significant pathological cardiac conditions (such as heart failure (NYHA class II-IV), conduction abnormalities (second- or third-degree AV block or sick sinus syndrome) not treated with a permanent pacemaker, significant ischemic heart disease or significant valvular disease.
PRILIGY is contraindicated for concomitant treatment with monoamine oxidase inhibitors (MAOIs), or within 14 days of discontinuing treatment with an MAOI. Similarly, an MAOI should not be administered within 7 days after PRILIGY has been discontinued (see PRECAUTIONS - Interactions with Other Medicines).
PRILIGY is contraindicated for concomitant treatment with thioridazine, or within 14 days of discontinuing treatment with thioridazine. Similarly, thioridazine should not be administered within 7 days after PRILIGY has been discontinued (see PRECAUTIONS - Interactions with Other Medicines).
PRILIGY is contraindicated for concomitant treatment with serotonin reuptake inhibitors [selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors (SNRIs), tricyclic antidepressants (TCAs)] or other medicinal/herbal products with serotonergic effects [e.g., L-tryptophan, triptans, tramadol, linezolid, lithium, St. John's Wort (Hypericum perforatum)] or within 14 days of discontinuing treatment with these medicinal/herbal products. Similarly these medicinal/herbal products should not be administered within 7 days after PRILIGY has been discontinued (see PRECAUTIONS - Interactions with Other Medicines).
PRILIGY is contraindicated for concomitant treatment with potent CYP3A4 inhibitors such as ketoconazole, itraconazole, ritonavir, saquinavir, telithromycin, nefazodone, nelfinavir, atazanavir, etc. (see PRECAUTIONS - Interactions with Other Medicines).
PRILIGY is contraindicated in patients with moderate and severe hepatic impairment.
PRECAUTIONS Patients must not take more than one tablet once every 24 hours due to increased risk of side effects (see Boxed Warning) and lack of additional benefit. PRILIGY is only indicated in men with PE. Safety has not been established and there are no data on the ejaculation-delaying effects in men without PE.
Use with recreational drugs Patients should be advised not to use PRILIGY in combination with recreational drugs. Recreational drugs with serotonergic activity such as ketamine, methylenedioxy-methamphetamine (MDMA) and lysergic acid diethylamide (LSD) may lead to potentially serious reactions if combined with PRILIGY. These reactions include, but are not limited to, arrhythmia, hyperthermia, and serotonin syndrome. Use of PRILIGY with recreational drugs with sedative properties such as narcotics and benzodiazepines may further increase somnolence and dizziness.
Ethanol Combining alcohol with PRILIGY may increase alcohol-related neurocognitive effects and may also enhance neurocardiogenic adverse events such as syncope, thereby increasing the risk of accidental injury; therefore, patients should be advised to avoid alcohol while taking PRILIGY (see PRECAUTIONS - Interactions with Other Medicines).
The number and percentage of subjects with syncope (characterized as loss of consciousness) was greater in PRILIGY-treated subjects than in those who were treated with placebo. A dose-response relationship for syncope is suggested based on subject incidence across all studies. In Phase 3 studies involving 6081 randomized subjects, the frequency of syncope characterised as a loss of consciousness was 0.06% for PRILIGY 30 mg and 0.05% for placebo, compared with a higher rate of 0.23% for an unapproved dose (60 mg) and 0.64% for all Phase 1 doses combined (30 mg to 240 mg) in Phase 1 non-PE subjects studies. In Phase 3 studies, three cases of syncope with bradycardia and sinus arrest (2 events, 5 seconds each; one event 28 seconds in duration) were observed during Holter monitor recording. Each subject spontaneously recovered normal sinus rhythm.
Possibly prodromal symptoms such as nausea, dizziness/light-headedness, and diaphoresis were reported more frequently among patients treated with PRILIGY compared to placebo. In patients receiving 30 mg PRILIGY in Phase 3 clinical trials, nausea was reported in 11.0%, dizziness in 5.8% and hyperhidrosis/diaphoresis in 0.8%.
Syncope observed in the clinical trials were considered vasovagal in etiology and the majority occurred during the first 3 hours after dosing, after the first dose, or associated with study- related procedures in the clinic setting (such as blood draw and orthostatic maneuvers and blood pressure measurements). Possibly prodromal symptoms, such as nausea, dizziness, light-headedness, palpitations, asthenia, confusion and diaphoresis generally occurred within the first 3 hours following dosing and often preceded the syncope. Patients need to be made aware that they could experience syncope at any time with or without prodromal symptoms during their treatment with PRILIGY. Prescribers should counsel patients about the importance of maintaining adequate hydration and about how to recognize prodromal signs and symptoms to decrease the likelihood of serious injury associated with falls due to loss of consciousness. If the patient experiences possibly prodromal symptoms, the patient should immediately lie down so his head is lower than the rest of his body or sit down with his head between his knees until the symptoms pass, and be cautioned to avoid situations where injury could result, including driving or operating hazardous machinery, should syncope or other CNS effects occur (see PRECAUTIONS - Effects on Ability to Drive and Operate Machinery).
Combining alcohol with PRILIGY may enhance neurocardiogenic adverse events such as syncope, thereby increasing the risk of accidental injury; therefore, patients should be advised to avoid alcohol with taking PRILIGY.
Subjects with underlying cardiovascular disease were excluded from Phase 3 clinical trials. The risk of adverse cardiovascular outcomes from syncope (cardiac syncope and syncope from other causes) is increased in patients with underlying structural cardiovascular disease (e.g., documented outflow obstruction, valvular heart disease, carotid stenosis and coronary artery disease). There are insufficient data to determine whether this increased risk extends to vasovagal syncope in patients with underlying disease.
Alteration in cardiac conduction was observed only in preclinical isolated in vitro/ ex vivo models, and not in whole-animal testing. In clinical studies, supraventricular beats and arrhythmias were slightly higher on PRILIGY. A review of Holter data in over 3350 subjects in Phase 3 clinical trials demonstrated asymptomatic non-sustained ventricular tachycardia occurring in 0.2%, 0.3%, and 0.3% of placebo, PRILIGY 30 mg and PRILIGY 60 mg dose groups, respectively. In addition, no effect on QTc prolongation was detected in two Phase I thorough QT studies designed to investigate the effect of PRILIGY on cardiac repolarization.
An orthostatic test should be performed before initiating therapy. In case of a history of documented or suspected orthostatic reaction, treatment with PRILIGY should be avoided.
Orthostatic hypotension has been reported in clinical trials. The prescriber should counsel the patient in advance that if he experiences possibly prodromal symptoms, such as light- headedness soon after standing, he should immediately lie down so his head is lower than the rest of his body or sit down with his head between his knees until the symptoms pass. The prescriber should also inform the patient not to rise quickly after prolonged lying or sitting. In addition, PRILIGY should be prescribed with caution in patients taking medicinal products with vasodilatation properties (such as alpha adrenergic receptor antagonists, nitrates, PDE5 inhibitors) due to possible reduced orthostatic tolerance (see PRECAUTIONS - Interactions with Other Medicines).
Caution is advised in all patients concomitantly treated with moderate CYP3A4 inhibitors. Unless the patient is known to be a CYP2D6 extensive metaboliser, caution is advised when used concomitantly with moderate CYP3A4 inhibitors (see DOSAGE AND ADMINISTRATION and INTERACTIONS WITH OTHER MEDICINES).
Caution is advised in patients taking potent CYP2D6 inhibitors or in patients known to be of CYP2D6 poor metaboliser genotype, as this may increase exposure levels, which may result in a higher incidence and severity of dose dependent adverse events (see DOSAGE AND ADMINISTRATION and INTERACTIONS WITH OTHER MEDICINES).
SSRIs have been shown to increase the risk compared to placebo of suicidal thinking and suicidality in short-term studies in children and adolescents with Major Depressive Disorder and other psychiatric disorders. Short-term studies did not show an increase in the risk of suicidality with antidepressants compared to placebo in adults beyond age 24. In clinical trials with PRILIGY for the treatment of premature ejaculation, there was no clear indication of treatment-emergent suicidality.
PRILIGY should not be used in patients with a history of mania/hypomania or bipolar disorder and should be discontinued in any patient who develops symptoms of these disorders.
Due to the potential of SSRIs to lower the seizure threshold, PRILIGY should be discontinued in any patient who develops seizures and avoided in patients with unstable epilepsy. Patients with controlled epilepsy should be carefully monitored.
Use in children and adolescents under age 18
PRILIGY should not be used in individuals below 18 years of age.
Co-morbid depression and psychiatric disorders
Men with underlying signs and symptoms of depression should be evaluated prior to treatment with PRILIGY to rule out undiagnosed depressive disorders. Concomitant treatment of PRILIGY with antidepressants, including SSRIs and SNRIs, is contraindicated (see CONTRAINDICATIONS). Discontinuation of treatment for ongoing depression or anxiety in order to initiate PRILIGY for the treatment of PE is not recommended. PRILIGY is not indicated for psychiatric disorders and should not be used in men with these disorders, such as schizophrenia, or in those suffering with co-morbid depression, as worsening of symptoms associated with depression cannot be excluded. This could be the result of underlying psychiatric disorder or might be a result of medicinal product therapy. Physicians should encourage patients to report any distressing thoughts or feelings at any time and if signs and symptoms of depression develop during treatment, PRILIGY should be discontinued.
There have been reports of bleeding abnormalities with SSRIs. Caution is advised in patients taking PRILIGY, particularly in concomitant use with medicinal products known to affect platelet function (e.g., atypical antipsychotics and phenothiazines, most tricyclic antidepressants [TCAs], acetylsalicylic acid, nonsteroidal anti-inflammatory drugs [NSAIDs], anti-platelet agents) or anticoagulants (e.g., warfarin), as well as in patients with a history of bleeding or coagulation disorders (see INTERACTION WITH OTHER DRUGS).
PRILIGY is not recommended for use in patients with severe renal impairment and caution is advised in patients with mild or moderate renal impairment (see DOSAGE AND ADMINISTRATION and PHARMACOKINETICS).
Abrupt discontinuation of chronically administered SSRIs used to treat chronic depressive disorders has been reported to result in the following symptoms: dysphoric mood, irritability, agitation, dizziness, sensory disturbances (e.g., paraesthesias such as electric shock sensations), anxiety, confusion, headache, lethargy, emotional lability, insomnia, and hypomania.
However, a double-blind clinical trial in subjects with PE designed to assess the withdrawal effects of 62 days of daily or as needed dosing with 60 mg PRILIGY showed no evidence of withdrawal syndrome and little evidence of withdrawal symptoms with only a slightly higher incidence of mild or moderate insomnia and dizziness reported in subjects switched to placebo after daily dosing (see CLINICAL TRIALS). Consistent results were seen in a second double- blind clinical trial with a 24-week treatment phase of 30 and 60 mg doses as needed followed by a 1-week withdrawal assessment period.
As with other SSRIs, the use of PRILIGY has been associated with ocular effects such as mydriasis and eye pain. PRILIGY should be used with caution in patients with raised intraocular pressure or those at risk of angle closure glaucoma.
In Phase 3 studies, PRILIGY 30 mg was associated with a higher incidence of mood related AEs, such as insomnia (2.2%), anxiety (1.1%) euphoric mood (0.2%), and depression (0.2%) compared to placebo (1.5%, 0.5%, 0%, 0.3%, respectively). Men with psychiatric disorders such as depression and anxiety were excluded from Phase 3 clinical studies. PRILIGY should not be used in men with concomitant psychiatric disorders, as it is unknown if the risk of these events and/or the underlying psychiatric condition could worsen (see PRECAUTIONS).
In Phase 3 studies, PRILIGY 30 mg was associated with a higher incidence of sexual AEs, such as erectile dysfunction (2.3%) and libido decreased (0.5%) compared to placebo (1.6% and 0.3%, respectively). Men with clinically significant erectile dysfunction and other sexual disorders were excluded from clinical studies. Caution should be used when prescribing PRILIGY to men with other forms of sexual dysfunction, as it is unknown if the sexual dysfunction could worsen.
In Phase 3 studies, PRILIGY 30 mg was associated with a higher incidence of accidental injuries (2.7%) compared to placebo (1.8%). While some accidents were reported to be related to syncope occurrence, there was no clear correlation between accidental injury adverse events and events of dizziness or changes in alertness associated with PRILIGY administration. Patients should be cautioned to avoid situations where injury could result should syncope or symptoms such as dizziness occur (see PRECAUTIONS, Effect on Ability to Drive or Operate Machineryand DOSAGE AND ADMINISTRATION).
No effects on fertility were observed in male rats given dapoxetine HCl at up to 0.25% of the diet, corresponding to a dose of 158 mg/kg/day. Plasma concentrations of dapoxetine in the animals did not reach clinical levels. As such, the potential for effects on fertility in patients receiving PRILIGY is not known.
Fertility was unaffected in female rats given dapoxetine at up to 100 mg/kg/day by oral gavage, yielding exposure to dapoxetine (plasma AUC) marginally higher than that of men at the maximum recommended human dose of 60 mg/day.
Use in Pregnancy Category C
PRILIGY is not indicated for use by women.
Dapoxetine was not teratogenic in rats at oral doses up to 100 mg/kg/day or in rabbits given up to 75 mg/kg/day during the period of organogenesis. Delayed ossification and an increased incidence of skeletal variations (extra ribs) were observed in fetuses at the highest doses tested, which were maternotoxic. Exposure to dapoxetine in rats (plasma AUC) was twice that of humans at the recommended clinical dose of 30 mg/day.
Dapoxetine and/or its metabolites cross the placenta in rats.
There are no adequate and well-controlled studies with dapoxetine in pregnant women. Neonates exposed to other SSRIs and SNRIs (serotonin and noradrenaline reuptake inhibitors) late in the third trimester have developed complications requiring prolonged hospitalisation, respiratory support and tube feeding. Such complications can arise immediately upon delivery. Reported clinical findings have included respiratory distress, cyanosis, apnoea, seizures, temperature instability, feeding difficulty, vomiting, hypoglycaemia, hypotonia, hypertonia, hyper-reflexia, tremor, jitteriness, irritability, lethargy, constant crying, somnolence and difficulty sleeping. These features are consistent with either a direct toxic effect of SSRIs and SNRIs or, possibly, a drug discontinuation syndrome.
There is no evidence to suggest that dapoxetine exposure has an effect on a partner's pregnancy based on limited observational data from the clinical trial database.
Use in Lactation
PRILIGY is not indicated for use by women.
It is not known if either dapoxetine or its metabolites are excreted in human breast milk.
Carcinogenicity
In studies with oral administration, dapoxetine was not carcinogenic to rats when administered daily for approximately two years at doses up to 225 mg/kg/day, yielding approximately three times the exposure to dapoxetine (plasma AUC) seen in human males given the recommended clinical dose of 30 mg. Dapoxetine also did not cause tumours in Tg.rasH2 mice when administered orally at the maximum possible doses of 100 mg/kg/day for 6 months and 200 mg/kg/day for 4 months. Exposure to dapoxetine in the mice at 100 mg/kg/day after 6-months administration was equivalent to that seen in humans at the 30 mg dose.
Daily topical dermal administration for 6 months to transgenic mice at 375, 750, or 1500 mg/kg/day produced some tumour promoter activity (papillomas at the application site) at 750 mg/kg/day or higher. Systemic exposure, to dapoxetine (plasma AUC) was approximately 2- to 3-fold that of males given the recommended clinical dose of 30 mg. The clinical relevance of this finding is unknown.
Genotoxicity
Dapoxetine was not mutagenic in vitro in the bacterial Ames assay or the forward mutation test in mouse lymphoma cells. Dapoxetine was not clastogenic in the in vitro chromosomal aberration test in Chinese hamster ovary cell or the in vivo mouse micronucleus assay. The major human metabolite, dapoxetine N-oxide, was negative in tests for bacterial mutagenicity and in vitro clastogenicity.
INTERACTIONS WITH OTHER MEDICINES
Potential for interaction with monoamine oxidase inhibitors
In patients receiving an SSRI in combination with a monoamine oxidase inhibitor (MAOI), there have been reports of serious, sometimes fatal, reactions including hyperthermia, rigidity, myoclonus, autonomic instability with possible rapid fluctuations of vital signs, and mental status changes that include extreme agitation progressing to delirium and coma. These reactions have also been reported in patients who have recently discontinued an SSRI and have been started on an MAOI. Some cases presented with features resembling neuroleptic malignant syndrome. Animal data on the effects of combined use of an SSRI and MAOIs suggest that these medicinal products may act synergistically to elevate blood pressure and evoke behavioural excitation. Therefore, PRILIGY should not be used in combination with an MAOI, or within 14 days of discontinuing treatment with an MAOI. Similarly, an MAOI should not be administered within 7 days after PRILIGY has been discontinued (see CONTRAINDICATIONS).
Potential for interaction with thioridazine
Thioridazine administration alone produces prolongation of the QTc interval, which is associated with serious ventricular arrhythmias. Medicinal products such as PRILIGY that inhibit the CYP2D6 isoenzyme appear to inhibit the metabolism of thioridazine and the resulting elevated levels of thioridazine are expected to augment the prolongation of the QTc interval. PRILIGY should not be used in combination with thioridazine or within 14 days of discontinuing treatment with thioridazine. Similarly, thioridazine should not be administered within 7 days after PRILIGY has been discontinued (see CONTRAINDICATIONS).
Medicinal/herbal products with serotonergic effects
As with other SSRIs, co-administration with serotonergic medicinal/herbal products (including MAOIs, L-tryptophan, triptans, tramadol, linezolid, SSRIs, SNRIs, lithium and St. John's Wort (Hypericum perforatum) preparations) may lead to an incidence of serotonin associated effects. PRILIGY should not be used concomitantly with other SSRIs, MAOIs, other serotonergic medicinal/herbal products or within 14 days of discontinuing treatment with these medicinal/herbal products. Similarly, these medicinal/herbal products should not be administered within 7 days after Priligy has been discontinued (see CONTRAINDICATIONS).
The use of PRILIGY in combination with CNS active medicinal products has not been systematically evaluated in patients with premature ejaculation. Consequently, caution is advised if the concomitant administration of PRILIGY and such medicinal products is required.
Effects of co-administered medicinal products on dapoxetine hydrochloride
In vitro studies in human liver, kidney, and intestinal microsomes indicate dapoxetine is metabolized primarily by CYP2D6, CYP3A4 and flavin monooxygenase 1 (FMO1). Therefore, inhibitors of these enzymes may reduce dapoxetine clearance.
CYP3A4 inhibitors - Potent CYP3A4 inhibitors
Administration of ketoconazole (200 mg twice daily for 7 days) increased the Cmax and AUCinf of
dapoxetine (60 mg single dose) by 35% and 99%, respectively. Considering the contribution of both unbound dapoxetine and desmethyldapoxetine, the Cmax of the active fraction may be
increased by approximately 25% and the AUC of the active fraction may be doubled if taken with potent CYP3A4 inhibitors.
The increases in the Cmax and AUC of the active fraction may be markedly increased in a part of
the population which lack a functional CYP2D6 enzyme, i.e., CYP2D6 poor metabolisers, or in combination with potent inhibitors of CYP2D6.
Therefore, concomitant use of PRILIGY and potent CYP3A4 inhibitors, such as such as ketoconazole, itraconazole, ritonavir, saquinavir, telithromycin, nefazodone, nelfinavir and atazanavir, is contraindicated (see CONTRAINDICATIONS). CYP3A4 inhibitors - Moderate CYP3A4 inhibitors
Concomitant treatment with moderate CYP3A4 inhibitors (e.g., erythromycin, clarithromycin, fluconazole, amprenavir, fosamprenavir, aprepitant, verapamil, diltiazem) may also give rise to significantly increased exposure of dapoxetine and desmethyldapoxetine, especially in CYP2D6 poor metabolisers. The maximum dose of dapoxetine should be 30 mg if dapoxetine is combined with any of these drugs (see PRECAUTIONS and DOSAGE AND ADMINISTRATION).
The Cmax and AUCinf of dapoxetine (60 mg single dose) increased by 50% and 88%,
respectively, in the presence of fluoxetine (60 mg/day for 7 days). Considering the contribution of both unbound dapoxetine and desmethyldapoxetine, the Cmax of the active fraction may be
increased by approximately 50% and the AUC of the active fraction may be doubled if taken with potent CYP2D6 inhibitors. These increases in the Cmax and AUC of the active fraction are
similar to those expected for CYP2D6 poor metabolisers and may result in a higher incidence and severity of dose dependent adverse events (see PRECAUTIONS).
The pharmacokinetics of dapoxetine (60 mg) in combination with tadalafil (20 mg) and sildenafil (100 mg) were evaluated in a single dose crossover study (C-2003-027). Tadalafil did not affect the pharmacokinetics of dapoxetine. Sildenafil caused slight changes in dapoxetine pharmacokinetics (22% increase in AUCinf and 4% increase in Cmax), which are not expected to
be clinically significant. However, PRILIGY should be prescribed with caution in patients who use PDE5 inhibitors due to possible reduced orthostatic tolerance (see PRECAUTIONS).
Effects of dapoxetine hydrochloride on co-administered medicinal products
Concomitant administration of single or multiple doses of 30 mg or 60 mg PRILIGY to patients receiving daily doses of tamsulosin did not result in changes in the pharmacokinetics of tamsulosin. The addition of PRILIGY to tamsulosin did not result in a change in the orthostatic profile and there were no differences in orthostatic effects between tamsulosin combined with either 30 or 60 mg PRILIGY and tamsulosin alone. However, PRILIGY should be prescribed with caution in patients who use alpha adrenergic receptor antagonists due to possible reduced orthostatic tolerance (see PRECAUTIONS). Medicinal products metabolized by CYP2D6
Multiple doses of dapoxetine (60 mg/day for 6 days) followed by a single 50 mg dose of desipramine increased the mean Cmax and AUCinf of desipramine approximately 11% and 19%,
respectively, compared to desipramine administered alone. Dapoxetine may give rise to a similar increase in the plasma concentrations of other drugs metabolized by CYP2D6. The clinical relevance is likely to be small.
Medicinal products metabolized by CYP3A
Multiple dosing of dapoxetine (60 mg/day for 6 days) decreased the AUCinf of midazolam (8 mg
single dose) by approximately 20% (range -60 to +18%). The clinical relevance of the effect on midazolam is likely to be small in most patients. The increase in CYP3A activity may be of clinical relevance in some individuals concomitantly treated with a medicinal product mainly metabolized by CYP3A and with a narrow therapeutic window. Medicinal products metabolized by CYP2C19
Multiple dosing of dapoxetine (60 mg/day for 6 days) did not affect the pharmacokinetics of a single 40 mg dose of omeprazole. Dapoxetine is unlikely to affect the pharmacokinetics of other CYP2C19 substrates.
Medicinal products metabolized by CYP2C9
Multiple dosing of dapoxetine (60 mg/day for 6 days) did not affect the pharmacokinetics or pharmacodynamics of a single 5 mg dose of glyburide. Dapoxetine is unlikely to affect the pharmacokinetics of other CYP2C9 substrates.
Medicinal products metabolized by CYP1A and 2B6
Repeated administration of dapoxetine induced CYP1A and 2B in laboratory animals. Dapoxetine may increase the clearance of substrates of CYP1A and 2B6.
In a single-dose crossover study, dapoxetine (60 mg) did not affect the pharmacokinetics of tadalafil (20 mg) or sildenafil (100 mg).
There are no data evaluating the effect of chronic use of warfarin with PRILIGY; therefore, caution is advised when PRILIGY is used in patients taking warfarin chronically. (See PRECAUTIONS) In a pharmacokinetic study, dapoxetine (60 mg/day for 6 days) did not affect the pharmacokinetics or pharmacodynamics (PT or INR) of warfarin following a single 25 mg dose.
Coadministration of a single dose of ethanol, 0.5 g/kg (approximately 2 drinks), did not affect the pharmacokinetics of dapoxetine (60 mg single dose) or the pharmacokinetics of ethanol, however, PRILIGY in combination with ethanol increased somnolence and significantly decreased self-rated alertness. Pharmacodynamic measures of cognitive impairment (Digit
Vigilance Speed, Digit Symbol Substitution Test) did not show a significant separation from placebo with either ethanol or PRILIGY alone but did show a statistically significant effect when PRILIGY was coadministered with ethanol versus ethanol alone. Concomitant use of alcohol and PRILIGY could increase the chance or severity of adverse reactions such as dizziness, drowsiness, slow reflexes, or altered judgment. Combining alcohol with PRILIGY may increase these alcohol-related effects and may also enhance neurocardiogenic adverse events such as syncope, thereby increasing the risk of accidental injury; therefore, patients should be advised to avoid alcohol while taking PRILIGY (see PRECAUTIONS). Effect on Ability to Drive or Operate Machinery
Dizziness, disturbance in attention, syncope, blurred vision and somnolence have been reported in subjects receiving dapoxetine in clinical trials. Therefore, patients should be warned to avoid situations where injury could result, including driving or operating hazardous machinery.
Combining alcohol with PRILIGY may increase alcohol-related neurocognitive effects and may also enhance neurocardiogenic adverse events such as syncope, thereby increasing the risk of accidental injury; therefore, patients should be advised to avoid alcohol while taking PRILIGY (see PRECAUTIONS - Interactions with other Medicines). ADVERSE REACTIONS
The safety of PRILIGY was evaluated in 6081 subjects with premature ejaculation who were randomised in five Phase 3 double-blind, placebo-controlled clinical trials. Of these subjects, 1616 received PRILIGY 30 mg as needed and 1612 received placebo. A total of 241 subjects were exposed to PRILIGY 30 mg for >121 days.
Syncope characterized as loss of consciousness has been reported in clinical trials and is considered medicinal product-related. The majority of cases occurred during the first 3 hours after dosing, after the first dose or associated with study-related procedures in the clinic setting (such as blood draw and orthostatic maneuvers and blood pressure measurements). Prodromal symptoms often preceded the syncope (see PRECAUTIONS).
Orthostatic hypotension has been reported in clinical trials (see PRECAUTIONS).
The most frequently reported AEs in subjects receiving PRILIGY included nausea, diarrhoea, dizziness and headache (Table 4). These AEs all occurred at higher frequency with doses of PRILIGY above 30 mg studied in phase 3 studies.
Treatment-Emergent Adverse Events (≥1%) by System Organ Class and Preferred Term in Phase 3 Placebo-Controlled Studies
(Dapoxetine SCS: Intent-to-Treat Analysis Set)
System Organ Class Total no. subjects with adverse events Gastrointestinal disorders
Diarrhoea (includes defaecation urgency)
Nervous system disorders
Dizziness (includes dizziness postural and dizziness exertional)
Infections and infestations Psychiatric disorders
Insomnia (includes middle insomnia and initial insomnia)
General disorders and administration site conditions Respiratory, thoracic and mediastinal disorders Investigations Musculoskeletal and connective tissue disorders Reproductive system and breast disorders
Adverse event counts (%) are based on the number of subjects, not the number of events. Studies included: C-2002-012, C-2002-013, R096769-PRE-3001 and R096769-PRE-3003.
The most common adverse drug reactions (5%) reported during clinical trials at a dose of 30 mg PRILIGY were headache, dizziness and nausea. The most common events leading to discontinuation were nausea (0.9% of 30 mg PRILIGY-treated subjects) and dizziness (0.7% of 30 mg PRILIGY-treated subjects).
Adverse drug reactions reported by PRILIGY-treated subjects in these trials are shown in Table 5.
Frequency of Adverse Drug Reactions Adverse Drug Reactions Uncommon System Organ common ( 1/100 to < 1/10) ( 1/1000 to < 1/100) ( 1/10000 to < 1/1000) (≥ 1/10) Psychiatric disorders
disorder, Bruxism, Euphoric mood, Indifference, Apathy, Mood altered, Initial insomnia, Middle insomnia, Anorgasmia, Confusional state, Hypervigilance, Thinking abnormal, Disorientation, Loss of libido
disorders
Depressed level of consciousness, Syncope, Syncope vasovagal, Dizziness postural, Akathisia
Eye disorders
PRECAUTIONS - Eye disorders), Visual disturbance
labyrinth disorders disorders Vascular disorders Respiratory, thoracic and mediastinal disorders Gastrointestinal Nausea Diarrhoea, disorders
Abdominal pain, Abdominal pain upper, Dyspepsia, Flatulence, Stomach discomfort, Abdominal distention
subcutaneous tissue disorders Reproductive
Erectile dysfunction Ejaculation failure,
system and disorders disorders and administration site conditions Investigations
Blood pressure diastolic increased, Blood pressure orthostatic increased
ADRs were determined from all clinical trial doses of dapoxetine, including those above 30 mg.
a Reported as the verbatim term "falls asleep quickly at bedtime."
Adverse drug reactions reported in the long-term open-label extension trial were consistent with those reported in the double-blind studies and no additional adverse drug reactions were reported.
Postmarketing Data
No newly identified adverse drug reactions have been reported during post-marketing use of PRILIGY.
DOSAGE AND ADMINISTRATION
For oral use. Tablets should be swallowed whole. It is recommended that tablets be taken with at least one full glass of water. Patients should be cautioned to avoid situations where injury could result should syncope or its prodromal symptoms such as dizziness or light-headedness occur (see PRECAUTIONS). PRILIGY has minor or moderate influence on the ability to drive and use machines. Dizziness, disturbance in attention, syncope, blurred vision and somnolence have been reported in subjects receiving PRILIGY in clinical trials. Therefore, patients should be warned to avoid situations where injury could result, including driving or operating hazardous machinery. Patients must not take more than one tablet once every 24 hours due to increased risk of side effects (see Boxed Warning) and lack of additional benefit.
The recommended dose for all patients is 30 mg, taken as needed approximately 1 to 3 hours prior to sexual activity.
The maximum recommended dosing frequency is once every 24 hours. PRILIGY may be taken with or without food (see PHARMACOKINETICS).
The physician who elects to prescribe PRILIGY for the treatment of premature ejaculation should evaluate the risks and patient-reported benefits of the medicinal product after the first four weeks of treatment or after 6 doses, whichever occurred earlier to assess the patient risk-benefit balance and to determine whether continuing treatment with PRILIGY is appropriate.
Safety and efficacy of PRILIGY have not been established in patients age 65 years and over as limited data are available in this population (see PHARMACOKINETICS).
PRILIGY should not be used in individuals below 18 years of age.
No dose adjustment is required but caution is advised in patients with mild or moderate renal impairment. PRILIGY is not recommended for use in patients with severe renal impairment (see PHARMACOKINETICS).
No dose adjustment is required in patients with mild hepatic impairment. PRILIGY is contraindicated in patients with moderate and severe hepatic impairment (Child-Pugh Class B and C) (see CONTRAINDICATIONS and PHARMACOKINETICS).
Known CYP2D6 poor metabolisers or patients treated with potent CYP2D6 inhibitors
Caution is advised in patients known to be of CYP2D6 poor metaboliser genotype or in patients concomitantly treated with potent CYP2D6 inhibitors (see PRECAUTIONS and PHARMACOKINETICS)
Patients treated with moderate or potent inhibitors of CYP3A4
Concomitant use of potent CYP3A4 inhibitors is contraindicated. Caution is advised when used concomitantly with moderate CYP3A4 inhibitors (see CONTRAINDICATIONS and PRECAUTIONS). OVERDOSAGE
There have been no reports of overdose during clinical trials.
There were no unexpected adverse events in a clinical pharmacology study of PRILIGY with daily doses up to 240 mg (two 120 mg doses given 3 hours apart). In general, symptoms of overdose with SSRIs include serotonin-mediated adverse reactions such as somnolence, gastrointestinal disturbances such as nausea and vomiting, tachycardia, tremor, agitation and dizziness.
In cases of overdose, standard supportive measures should be adopted as required. Due to high protein binding and large volume of distribution of dapoxetine hydrochloride, forced diuresis, dialysis, hemoperfusion and exchange transfusion are unlikely to be of benefit. No specific antidotes for PRILIGY are known.
Contact the Poisons Information Centre (telephone 13 11 26) for advice on management of overdosage.
PRESENTATION AND STORAGE CONDITIONS
PRILIGY Tablets are round, film-coated tablets and are available in packs of 3, 6 or 18 tablets. The 18 tablets pack size is not currently marketed.
30 mg: light grey tablets debossed with "30" inside a triangle on one side.
PRILIGY tablets should be stored below 25°C.
NAME AND ADDRESS OF THE SPONSOR
A. Menarini Australia Pty Ltd Level 8, 67 Albert Ave Chatswood, NSW 2067.
POISON SCHEDULE OF THE MEDICINE DATE OF FIRST INCLUSION IN THE AUSTRALIAN REGISTER OF THERAPEUTIC GOODS (THE ARTG) DATE OF MOST RECENT AMENDMENT
PRILIGY is a registered trademark of ORTHO-MCNEIL PHARMACEUTICAL for dapoxetine tablets
02.00u, er begint wat te rommelen. Geen onweer, tenzij in mijn darmen. Een eerste bezoek aan het kleinste kamertje dringt zich op en het zal niet het laatste zijn. Veel slapen is niet van toepassing deze nacht en ik verschijn dan ook als een halve zombie aan de ontbijttafel. Een stevig ontbijt zit er zeker niet in, enkel een tas koffie om mijn pillen imodium door te slikken. Een normaal men
Loomade mikroobide antibiootikumiresistentsuse monitooring SISSEJUHATUS. Põllumajandusministeeriumi rakendusuuringute programmi toetusel on loomade mikroobide antibiootikumiresistentsuse uuringuid tehtud alates 2001 aastast. Alates 2006 aastast on mikroobide antibiootikumiresistentsuse monitooringus kasutatud MIC-metoodikat(VetMIC™, Swedish National Veterinary Institute,Uppsala
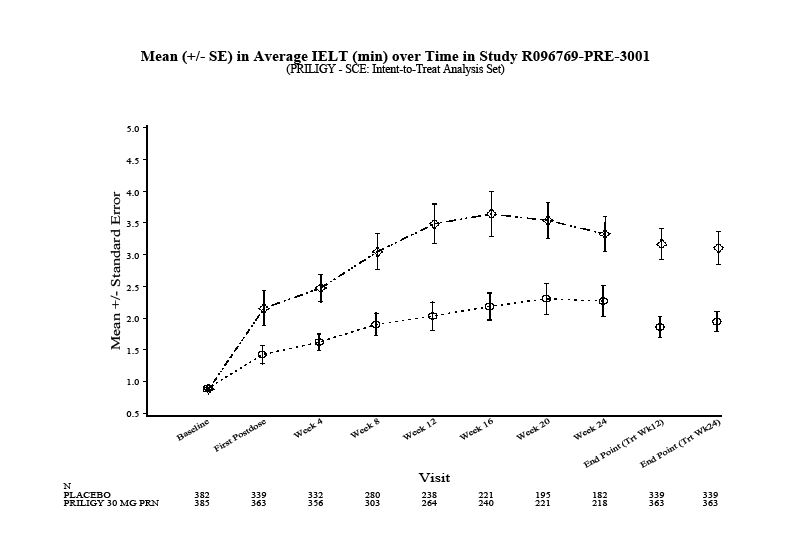 Figure 1: Study R096769-PRE-3001
Figure 1: Study R096769-PRE-3001